Internal Transcribed Spacers (ITS) are region within the ribosomal transcript that are excised and degraded during maturation. Their sequences generally show more variation than the ribosomal sequence, making them popular for phylogenetic analysis and/or identification of species and strains. Especially for fungi, this is a popular technique for identification as identification based on morphological features is laborious and often does not lead to a correct result. The region used is usually a combination of ITS and ribosomal sequences and most often consists of a (partial) 18S rRNA sequence (16S rRNA for prokaryotes), an internally transcribed region (ITS1), the whole sequence of 5.8s rRNA, an internally transcribed region (ITS2) and a (partial) sequence of 28s rRNA. Many ITS sequences are available on public databases such as NCBI and EBI.
ITS typing workflow in the BIONUMERICS software
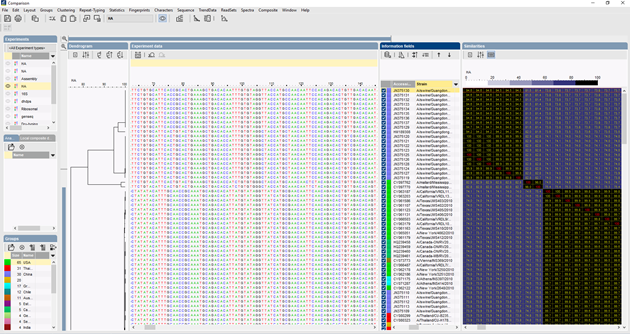
In BIONUMERICS, the ITS sequences can be imported in batch from trace files using the batch sequence assembly plugin. Reference sequences can be imported from public databases and/or from fasta or GenBank files.
Several algorithms can be used to perform a multiple alignment of the sequences, Needleman-Wunsch, Wilbur-Lipman and our own proprietary algorithm. Manual adaptation of the alignment is possible. Based on the multiple alignment, a clustering can be calculated to reflect the phylogenetic relationships between the analyzed sequences.
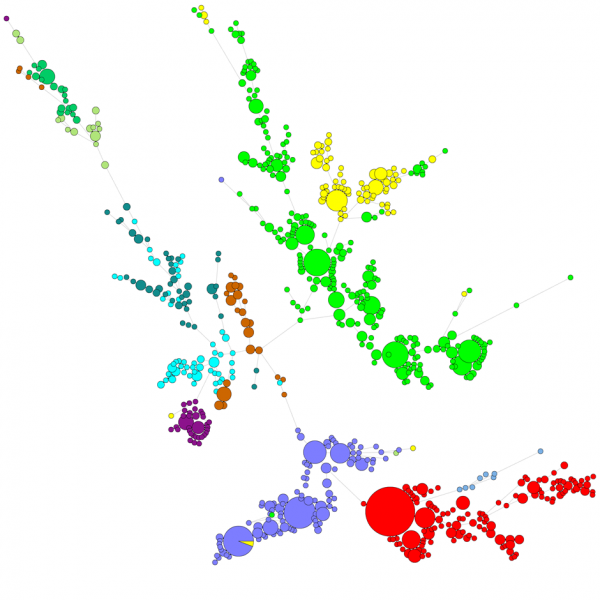
When comparing a large number of sequences, a UPGMA or neighbor-joining dendrogram does not give a very good overview of the data. In these cases, an unrooted tree provides a better solution, in BIONUMERICS it is possible to calculate minimal spanning trees, maximum likelihood trees and maximum parsimony trees starting from sequence data. Different groups can be defined for the data and these groups are visualized in the trees, giving a nice overview of the phylogenetic relationships, the cluster present in the database and also the outliers. The figure below shows a minimal spanning tree based on ITS sequences, the groups represent different genera.
The ITS sequence can also be used to identify unknown organisms against a database. With the Classifiers and Identification module, the user can create identification projects containing identified strains and use these to identify an unknown strain, a score will be assigned based on the similarity together with the reliability of this score. The user has full control over the parameters for comparison and the cutoff values for identification.